AI4Life Bartering Corner
Welcome to the AI4Life Bartering Corner! This is a space to foster collaboration between computational experts and life scientists.
Not all the projects could receive support from the AI4Life expert team, but we are still committed to providing the opportunity for the two communities to connect. This is the primary goal of the AI4Life Bartering Corner.
- Life scientists: besides the Bartering Corner, Euro-BioImaging Nodes also offer support, please refer to the Euro-BioImaging Web Portal for more information.
- Computational experts: this space is for you! Get in touch with the project you are interested in through image.sc.
Project 1: Analysis of morphological phenotypic aspects of 3D mouse embryos through micro-CT acquisitions
CONTACT: rita88
Challenge

I would like to identify, in an automatic manner, the sex of embryos and quantify several morphological parameters, such as heart or liver volumes.
DATA
The images show acquisitions of mouse embryos through micro-computed tomography
Project 2: Quantification of infection parameters in cell lines infected with Trypanosoma cruzi
CONTACT: victoria_alonso1
Challenge
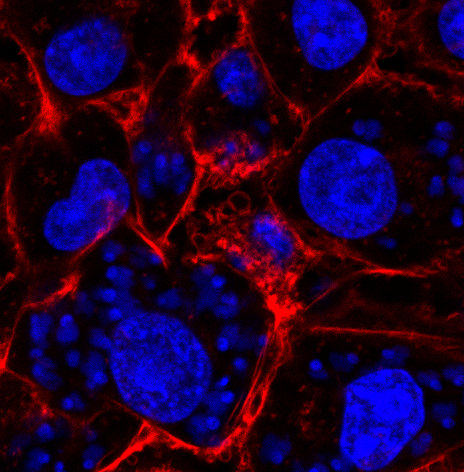
We are currently quantifying the number of infected cells and the average number of parasites in each infected cell by counting manually but it would be game-changing to have a tool to automatize this process. I have tried some image analysis tools but haven’t been able to achieve this goal just yet. We can provide images of the infected cells in different conditions, using different T. cruzi strains and with different markers. We have fluorescence markers as well as infected cells stained with May Grünwald-Giemsa staining, a classic histopathology staining protocol.
We need to obtain these parameters from the images: number of infected cells (with amastigotes inside) and total number of cells. From the infected cells the number of parasites inside each cell.
DATA
Trypanosoma cruzi, the etiological agent of Chagas disease or American trypanosomiasis, is a parasite with a complex life cycle that alternates between mammalian and insect hosts (Triatominidae family) that serve as the biological vector of this disease. T. cruzi life cycle can be reproduced completely in axenic conditions under Biosafety regulations. The intracellular stage of this parasite, which is called amastigote is the replicative form inside the mammalian host. Amastigotes infect almost all types of nucleated cells and have a tropism for cardiac muscle for example. This is what contributes to the clinical manifestations of the disease that can eventually lead to the death of the infected persons. We have a standardized protocol to infect culture cell monolayers in a 24 wells format which is then subject to fluorescence microscopy or bright field microscopy to stain the nucleus of the host cells and of the parasites infecting these cells. We also have other markers to identify only the parasite`s cytoskeleton and only the host cell membrane. It is important to know that amastigotes have a nucleus in the central region and a rod-shaped kinetoplast in the anterior area of the cell body, this structure also is composed of DNA and stains with common DNA markers, which adds a layer of complexity to the image analysis.
Image2 is a confocal image stained with phaloidine (red) that marks the actin cytoskeleton of the host cell. In blue we mark the DNA of the host cells (big nucleus) and the DNA of the parasite (small nucleus and kinetoplast).
Image3 and Image4 are also infections but stained with a dye that allows for bright-field microscopy. Again we see in light gray the cytoplasm of the host cell and darker the DNA from the host cell and the parasite.
Image2
Image3
Image4
Project 3: Spatial organization of polytene chromosomes in nuclei of Drosophila
CONTACT: SlovDro
Challenge
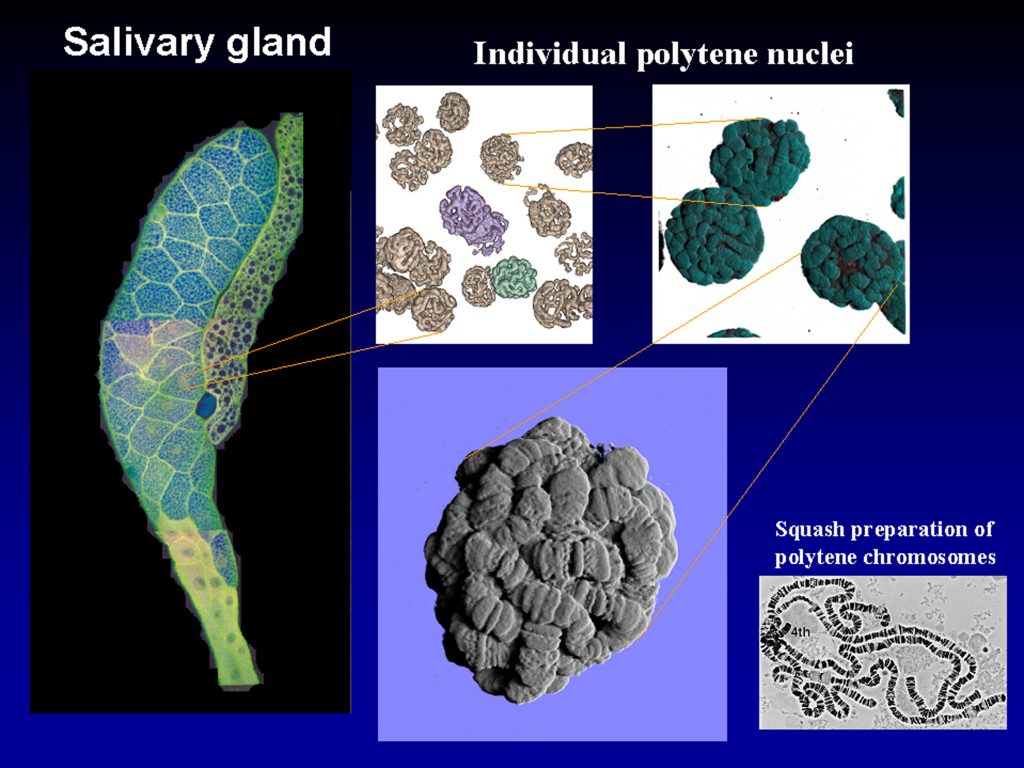
Drosophila polytene chromosomes are highly condensed chromosomes in permanent somatic conjugation, they do not divide in classical way as diploid chromosomes, they continuously grow in size by endoreplication, and attain 2000-fold level of ploidy in comparison to diploid cells. This situation requires specific arrangement of these giant chromosomes inside nuclei, and still allowing them to be biologically active in continued replication and very intense transcription. They are characterized by typical banding pattern which was a prototype model served for analysis and construction of the first highly detailed cytogenetical maps as early as in 1930s.
In spite of these facts, spatial (3D) organization of the polytene chromosomes is not resolved, and we do not know yet how individual chromosomal arms are packed to produce evident and tightly compacted ball. We still continue in scanning further nuclei, and we are aimed to understand how chromosomes are organized. As one of the first steps towards this goal we would like to unwide or unmount individual polytene chromosomes which then can be easily identified by individual loci according to above mentioned cytogenetic maps. These cytogenetic maps were produced by studying classic squash preparation where, however, no spatial organization and relationships between chromosomal arms can be recognized.
We expect that these goals will require segmentation of Z-stack images that would then also allow for reconstruction of this tight chromosomal pack via reverse flow. Very important moment will be to identify key steps in unmounting and reverse flow of this reconstruction strategy that will facilitate not only identification of the position of individual arms inside nucleus but also recognition of the points or foci where arms or their specific loci can interact with their partnering loci to form ordered organization of polytene nucleus.
DATA
Image data show selected single-plane sections of Z-scanned polytene chromosomes as they are packed inside nuclei of Drosophila salivary glands. Images were obtained using Zeiss LSM880-Airyscan2 microscope at 100x objective lenses. Staining dye was Hoechst 33258 binding to DNA, and excitation laser was 405 nm. Data files are in *.czi format, ZEN joint-deconvolved and Airyscan processed. We are looking for collaboration with IT-experts to perform advanced image analysis, most probably with segmentation software.
Project 4: AI for detecting invasive species
CONTACT: annaunderwater
Challenge
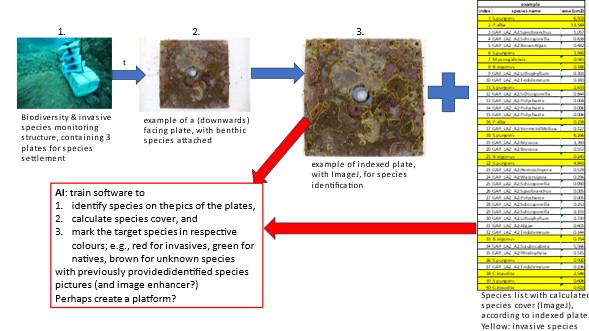
Why is this important? Until today there is no international standardized monitoring method for marine biodiversity or the presence of invasive species. Monitoring plates are nowadays quite common (see SERC-protocoll or Autonomous Reef Monitoring Systems, ARMS), but these methods are quite time-consuming. If we could use AI to at least pre-identify the species found on the structures/plates, it could save us loads of time, giving us more space for more monitoring in different places.
Why targeting invasive species? These are species which are introduced anthropogenically to different places, causing economic loss (damage on e.g. ship hulls), ecological disturbance (e.g. they outcompete native species, impact food chain) or carry pathogens which are harmful for humans.
DATA
This project aims to find a suitable monitoring method for measuring marine biodiversity and to determine the presence of invasive species (species that were introduced by humans, e.g., through ship traffic or recreational activities). Monitoring structures (see image1) consisting of 3 PVC plates were deployed in different locations to let species settle on the plates. As the locations differed in levels of anthropogenic disturbance and pollution, the community compositions differ among the locations in relation to (invasive) species presence, diversity, and species cover. After 3 months, structures were retrieved, the plates disassembled with attached and grown benthic organism (image2), and the species found were identified (image 3), species cover was calculated (with ImageJ), and a species list was created (image 4). All the species of all the plates and locations are already identified.
In order to speed up the process of species identification, my idea was to train a software (for instance, DeepImageJ?) to distinguish between target species (in this case, invasive species) and non-target species (native species, unknown species). The target species on the list (image 3) are marked in yellow; however, there are more species to target 🙂
Ideally, the software would produce an image with the target species marked in red (invasive species), the other marked in green (for native, “normal” species) and perhaps another colour for the unidentified/unknown species.
This might work by providing and training the software with identified species pictures, so for each target species I would provide a certain amount of pictures, macro-and microscopic. Then I think an image enhancer would be needed. With this trained skills the software should be able to distinguish between the target and non-target species.
Image1
Image2
Image3
Image4